






 您的位置 :
您的位置 :
一、标题:
直接热剥离法制备原子级分散的Co-P单元用于碱性氢电合成

二、论文相关信息:
第一作者:周正,苏怡心
通讯作者:赵慎龙
通讯单位:西南交通大学、国家纳米科学中心、日本东北大学
论文DOI:10.1021/jacs.4c11788
三、正文内容:
1、 全文速览:
本研究通过中温磷化剥离策略,在超薄碳纳米片上合成了原子分散的CoP4单元(CoP4−SSC),并深入探究其在碱性析氢反应(HER)中的性能。X射线吸收光谱(XAS)和理论计算揭示了Co−P单元的热力学形成机制。结果表明,CoP4−SSC在碱性HER中表现出优异性能:在10 mA cm-2电流密度下,过电位仅52 mV,转化频率(TOF)高达23.83 s-1。将其应用于阴离子交换膜(AEM)电解槽,仅需1.94 V电压即可实现1 A cm-2电流密度,并在500小时连续电催化过程中几乎无性能衰减。原位表征与密度泛函理论(DFT)计算表明,原子分散的Co−P单元构建了[P-*H···H2O*-Co]纳米界面,有效促进了Volmer-Heyrovsky路径中的水解离过程,为AEM电解槽的商业化提供了高效稳定的析氢电催化剂,开辟了新的应用方向。
2、 背景介绍:
碱性析氢反应(HER)在AEM电解槽中至关重要,直接影响绿色氢能的规模化生产。然而,碱性HER的反应动力学较酸性条件慢2-3个数量级,对催化剂的活性和稳定性提出了更高要求。尽管非贵金属电催化剂(如磷化物、硫化物、碳化物)在HER性能提升方面取得进展,但长期运行中活性与稳定性的矛盾仍未解决。近年来,基于MX4单元(X=C、N、O、P)的原子分散催化剂因催化活性位点和结构稳定性优势受到关注。研究表明,P的3p轨道与金属3d轨道形成的弱极性共价键可优化中间体吸附强度,显著提升HER性能;同时,P位点的低电负性使其与金属位点在水分解过程中形成协同活性中心。基于此,优化CoP4单元的形成和分布,构建新型碱性析氢催化剂,有望突破传统非贵金属催化剂的性能限制,为高效稳定的碱性HER提供新思路。
3、 研究亮点:
l 本研究采用中温磷化剥离策略,成功合成了原子分散的CoP4单元(CoP4-SSC),并均匀分布于超薄碳纳米片表面。该方法显著提高了活性位点的利用率,同时避免了传统合成中金属团聚的问题,为非贵金属催化剂的设计提供了新思路。
l CoP4-SSC在碱性析氢反应(HER)中表现出优异的性能。在10 mA cm-2电流密度下,其过电位仅为52 mV,转化频率(TOF)高达23.83 s-1,接近商业Pt/C催化剂。在实际应用测试中,以CoP4-SSC为阴极的阴离子交换膜(AEM)电解槽展现了优异性能。同时,其制氢成本仅为2.89美元/千克,显著低于市场平均水平,兼具经济性和可持续性。
l 原位表征与密度泛函理论(DFT)计算表明,CoP4单元通过形成[P-*H···H2O*-Co]纳米界面,显著促进了Volmer-Heyrovsky路径中的水解离过程。这种弱极性共价键特性在提升反应动力学和中间体吸附调控方面起到了关键作用。
4、 图文解析:
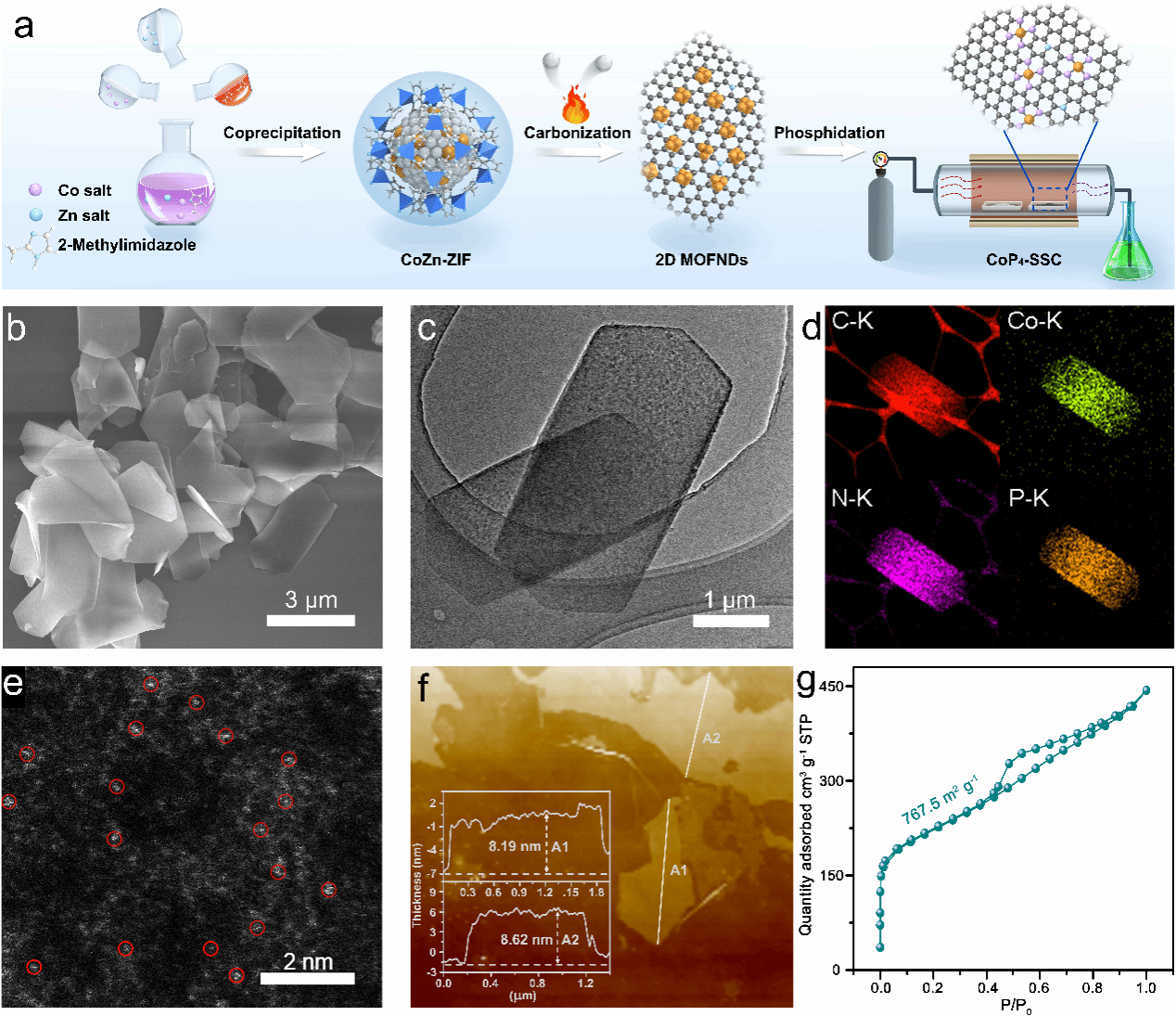
图1. 本文通过模板热解法结合中温磷化剥离技术制备了CoP4-SSC,SEM和HRTEM确认其为六边形超薄纳米片结构,
EDS和HAADF-STEM显示Co原子均匀分散,AFM测得厚度约8 nm,BET分析表明其具有大比表面积(767.5 m2 g-1),为高效电催化提供了优越结构基础。
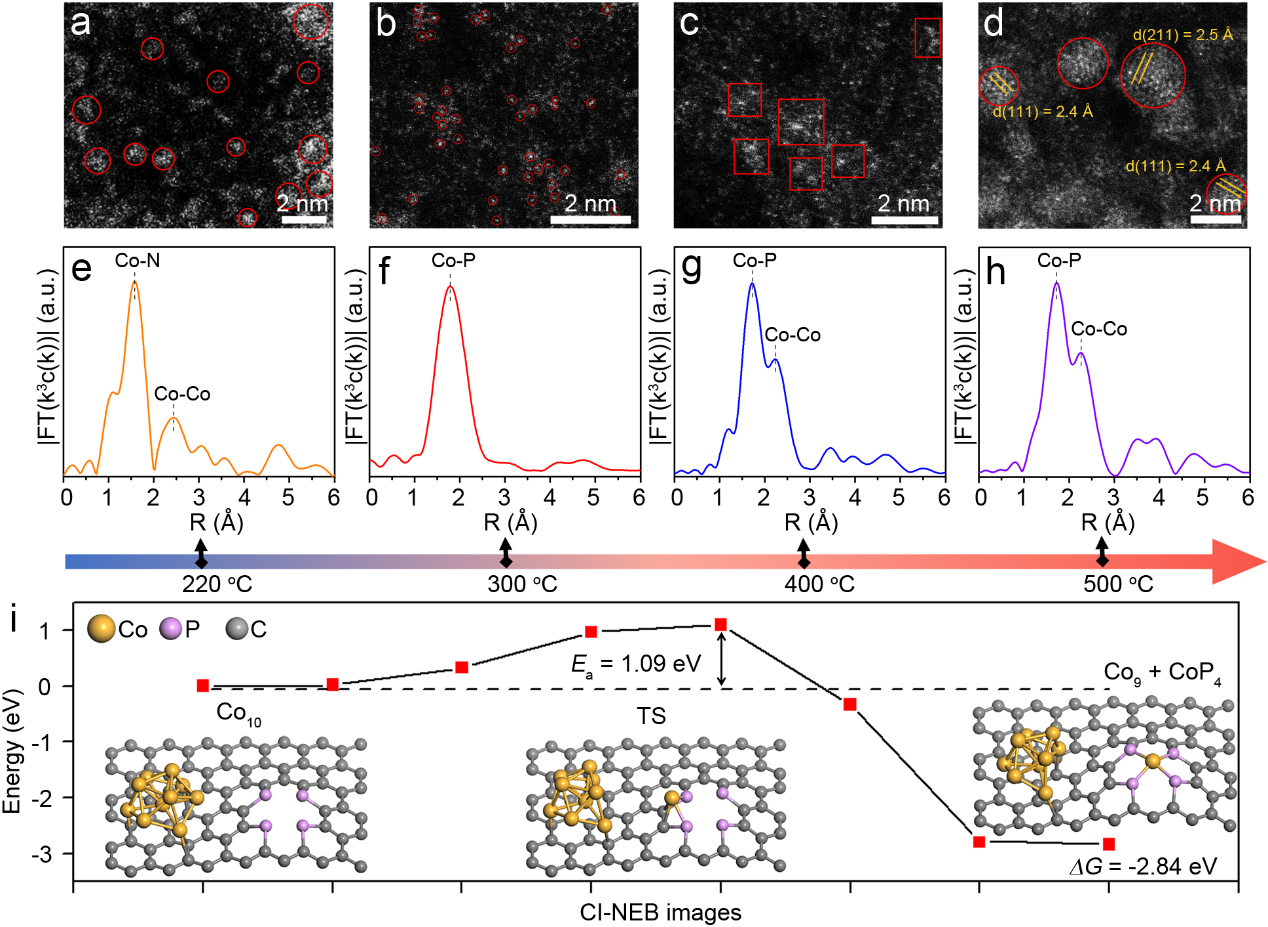
图2. 本文利用HAADF-STEM和FT-EXAFS表征分析了不同磷化温度对Co单原子位点剥离的热力学机制(图2)。结果显示,适中的磷化温度(300 °C)有助于形成稳定的CoP4单元,而更高温度(400-500 °C)则导致Co位点聚集并形成金属簇。此外,DFT计算表明,从Co簇中剥离单个Co位点的过程需要克服1.09 eV的能垒,并伴随2.84 eV的放热,这验证了剥离机制的可行性(图2i)。这些结果为优化单原子催化剂的设计提供了理论指导。

图3. 本文通过XANES和FT-EXAFS深入解析了CoP4−SSC的原子和电子结构。Co K边XANES光谱显示Co原子的价态小于+2,FT-EXAFS拟合确认了稳定的Co-P配位结构。C K边和P L边光谱表明P的掺入显著改变了基底的电子结构,形成了独特的Co-P键环境。电子密度差分图揭示,Co和P之间的电荷重分布降低了中间体的吸附能。PDOS分析显示CoP4-SSC的d轨道中心接近费米能级,与CoN4相比提供了更优的电子供体能力。这些结果表明Co-P键的协同作用在优化HER性能方面至关重要。
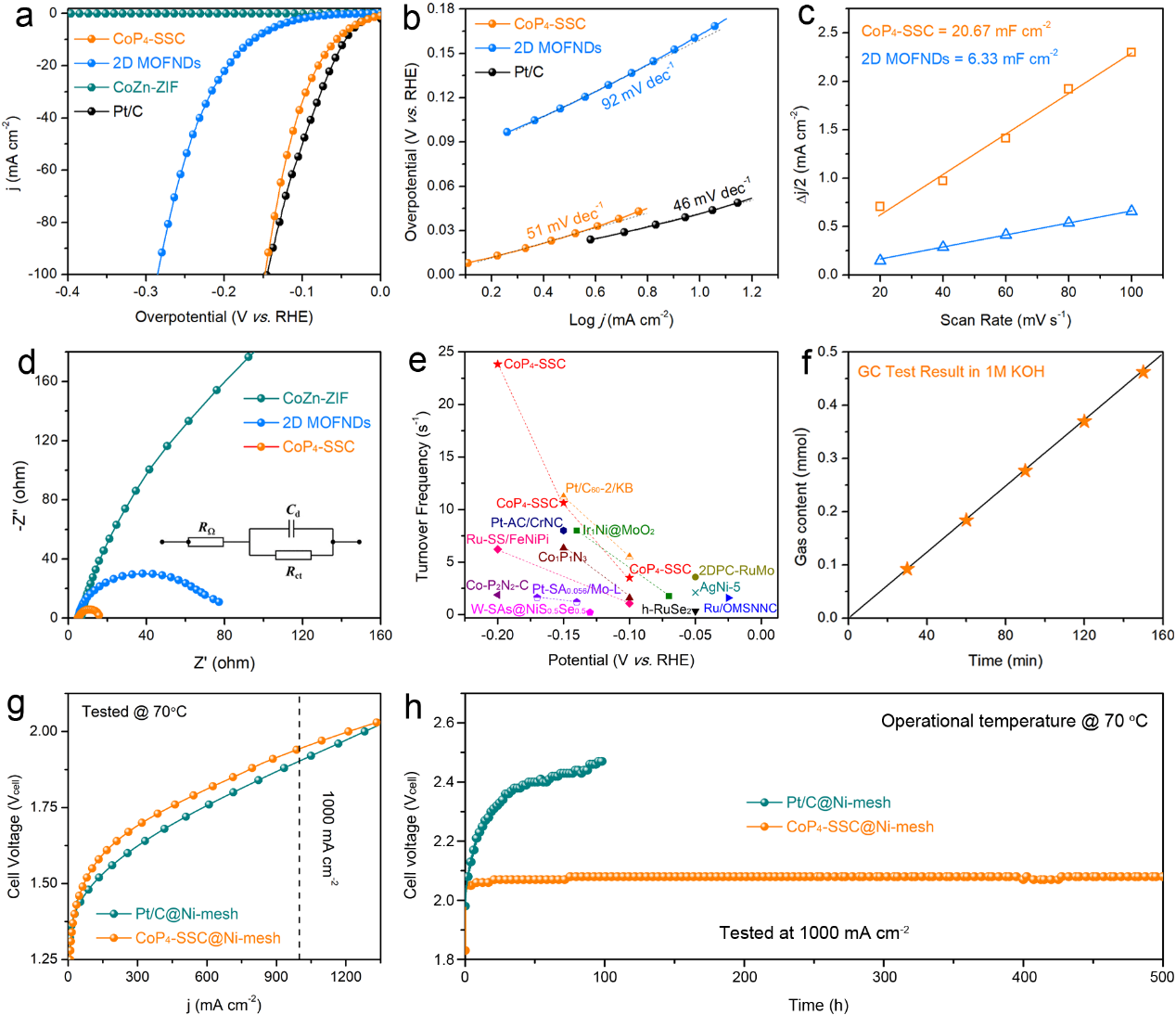
图4. 展示了CoP4-SSC在碱性介质中作为HER电催化剂的电化学性能。线性扫描伏安曲线(LSV)表明,CoP4-SSC在10 mA cm-2电流密度下的过电位仅为52 mV,显著优于对比样品2D MOFND的162 mV,接近商业Pt/C催化剂的表现(图4a)。塔菲尔斜率为51 mV dec-1,表明CoP4-SSC具有快速的反应动力学(图4b)。双电层电容(Cdl)测试显示,CoP4-SSC的Cdl值高达20.67 mF cm⁻²,远高于2D MOFNDs样品,表明其具有更大的电化学活性表面积(图4c)。电化学阻抗谱(EIS)进一步验证了CoP4-SSC的快速电荷转移能力,其电荷转移电阻(Rct)仅为10.7 Ω,显著低于参比样品(图4d)。此外,CoP4-SSC在不同过电位下的转化频率(TOF)高达23.83 s⁻¹,超越大多数现有HER电催化剂(图4e)。法拉第效率测量显示,CoP4-SSC的效率高达99.64%(图4f)。在阴离子交换膜(AEM)电解槽中的测试表明,采用CoP4-SSC作为阴极的电解槽在1 A cm-2电流密度下的工作电压仅为1.94 V,并在500小时运行中表现出优异的稳定性(图4g, h)。这些结果表明,CoP4-SSC在HER电催化中兼具高活性、快速动力学和良好稳定性,为其在工业级制氢中的应用奠定了基础。
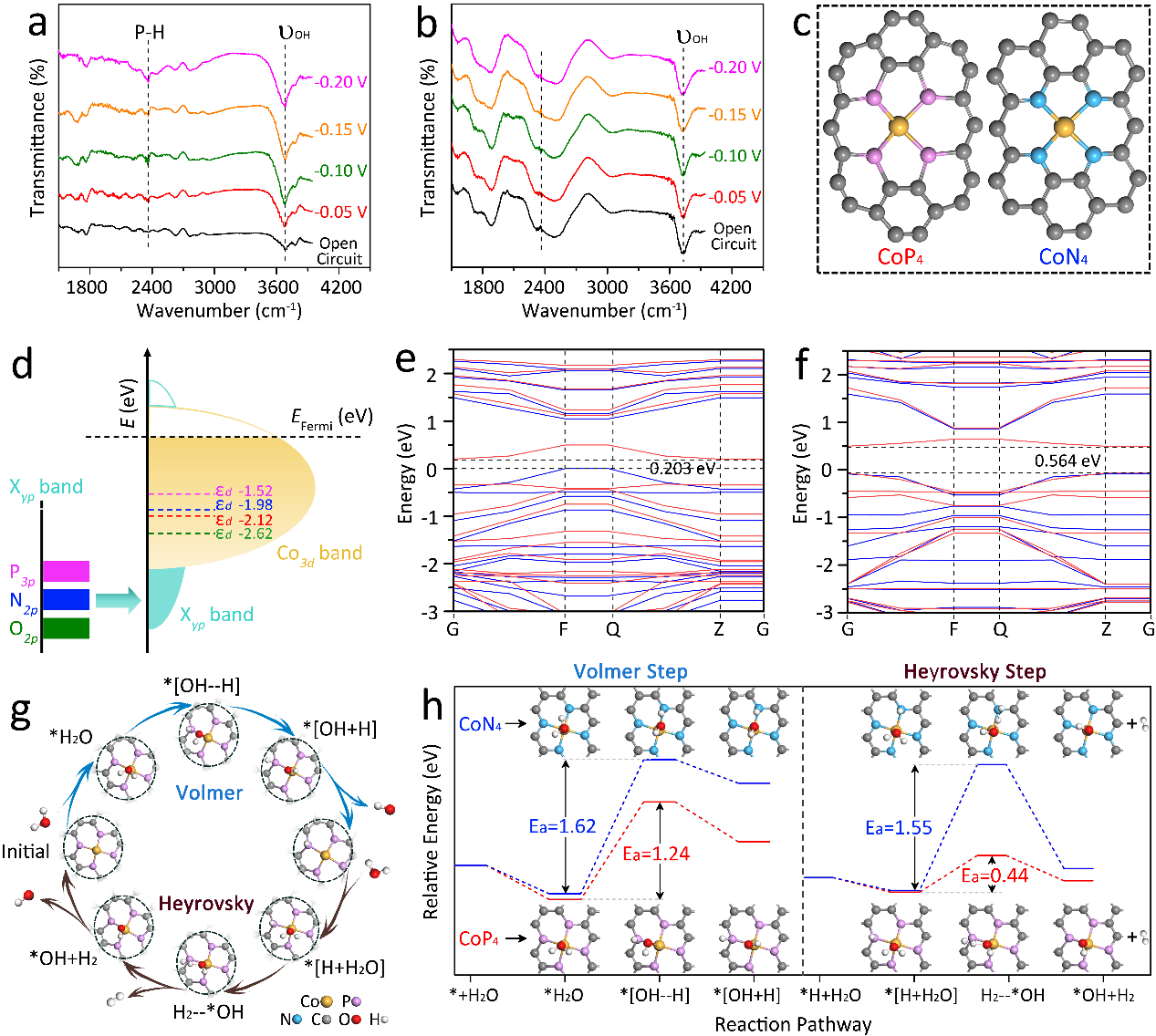
图5. 系统解析了CoP4-SSC在碱性析氢反应(HER)中的催化机理,结合原位表征和理论计算,揭示其优异性能的根源。原位SR-FTIR光谱显示,随着过电位的增加,CoP4-SSC中O−H和P−H特征峰强度显著增强,表明Co和P位点分别对羟基和*H物种具有协同吸附能力,有助于加速水分解和氢气释放。相比之下,商业CoP未表现出类似行为。通过DFT计算发现,CoP4-SSC的d带中心(−2.12 eV)优化至接近理想值,能够实现中间体吸附能的最佳平衡。此外,其Volmer和Heyrovsky路径的能垒分别为1.24 eV和0.44 eV,显著低于其他对比样品,显示出更低的反应能垒和更优的动力学性能。进一步提出的[P-*H···H2O*-Co]纳米界面模型表明,Co-P双位点通过协同催化作用,在提升反应活性和降低能垒方面具有关键作用。这些结果不仅阐明了CoP4-SSC高效HER性能的微观机制,也为设计新型单原子催化剂提供了理论依据和启示。
5、 总结与展望:
本文通过中温磷化剥离策略制备了热稳定的CoP4-SSC电催化剂,用于碱性析氢反应。XANES和FT-EXAFS高级表征表明,Co位点锚定在MOF衍生碳基体的四配位CoP4单元中。CoP4单元通过原位SR-FTIR分析和DFT计算揭示,能够有效激活第一配位壳的P位点参与催化,从而构建了动态优选的[P-*H···H2O*-Co]纳米界面。实验结果显示,CoP4-SSC在1 M KOH溶液中表现出优异的HER性能,10 mA cm-2电流密度下的过电位仅为52 mV,塔菲尔斜率为51 mV dec-1,转化频率TOF高达23.83 s-1,超越了最新报道的大多数非贵金属电催化剂。在阴离子交换膜AEM电解槽中,作为阴极电催化剂的CoP4-SSC仅需1.94 V即可驱动1 A cm-2的电流密度,且在500小时的连续运行中表现出优异的稳定性。该研究提出了一种激活非金属原子参与水解离的新策略,大幅提升了HER动力学性能,同时展示了单原子催化剂与AEM电解槽结合的高效性,为潜在工业应用奠定了基础。
